临床诊断“金标准”,qPCR的原理你都知道么?
qPCR全称是荧光定量PCR(Realtime fluorescence quantitative PCR),也称作实时荧光定量PCR,凭借其高精度、可定量等优势,是现阶段分子诊断主流技术平台,临床诊断的“金标准”,尤其广泛应用于感染性疾病(病毒性肝炎、性病和其他病菌/病毒类等)和肿瘤伴随诊断领域。然而,qPCR其中涉及的化学原理与数学原理大家都清楚么?今天,我们就和大家详细地聊一聊。
常规PCR技术
1983年,Kary Mullis在行驶在盘山公路的途中,突然想到DNA可以像盘山公路一样,一遍一遍地循环扩增,自此发明了PCR技术,也因此获得了诺贝尔奖。常规的PCR技术可以通过对终产物进行凝胶电泳来做定性分析和粗略的定量比对,但是无法对整个扩增反应进行实时监测,且无法对起始模板的进行准确定量。后来,实时荧光定量PCR技术的出现就满足了这样的需求。
荧光定量PCR
实时荧光定量PCR定义:
利用荧光信号的变化实时检测PCR扩增反应中每一个循环扩增产物量的变化,通过Ct值和标准曲线的关系对起始模板进行定量分析
1、化学原理
常用的荧光标记方法有两种:SYBR Green I 染料法和Taqman探针法
SYBR Green I 染料法
SYBR Green I 是一种结合于所有双链DNA(dsDNA)双螺旋小沟区域的具有绿色激发波长的染料。
当SYBR Green I 游离在体系中时,会发出微弱的荧光,当与DNA双链结合的时候,荧光信号会放大到几千倍,这时候就会被仪器检测到。在PCR的过程中,当进行到引物退火和延伸反应时,SYBR Green I 会逐渐地结合到DNA双链上,并且结合量与双链的浓度成正比,所以荧光信号的强度能反映出体系中双链DNA的浓度。
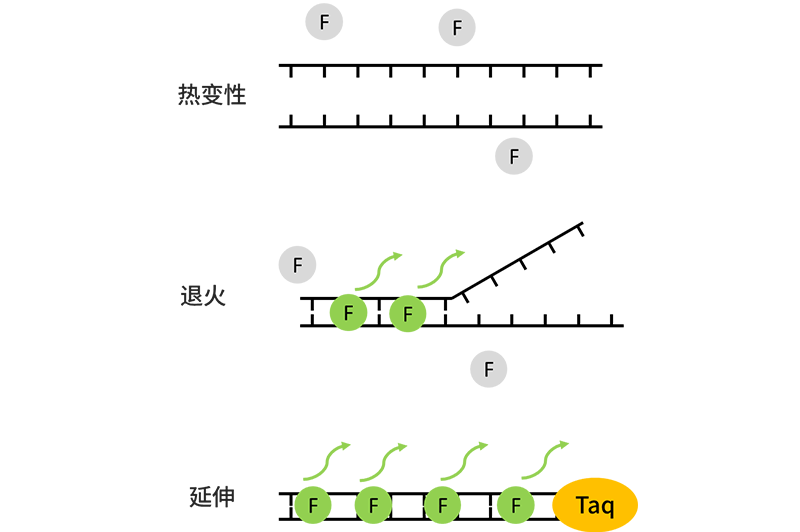
染料法操作简单、灵敏度高且成本较低,被广泛地用于科研中对多个目的基因定量分析和基因表达量的研究。但由于SYBR Green I 是一种非特异性荧光标记,如果反应体系中具有非特异性扩增和引物二聚体,也会被检测到荧光信号,因而会对检测结果产生影响。实验中我们通常需要重新设计引物,来防止产生非特异性扩增。
那么在实验中要如何判断体系是否有非特异性扩增呢?
这就要用到另一个概念——熔解曲线

当PCR循环反应完成之后,系统会测定熔解曲线,通常将体系从60℃加热到90℃。然后每隔一定时间测定荧光强度,随着温度升高,dsDNA双链解开,SYBR Green I 染料脱落,荧光值逐渐下降,就形成了一个温度和荧光强度的曲线。当加热到一定温度的时,曲线会陡降,这个温度就是Tm值。我们将荧光强度对温度的变化求导,就得到了我们常见的带峰的熔解曲线。每一个峰就代表一种特异性产物,所以通过峰的形状和多少就可以判断体系中是否存在引物二聚体和非特异性结合。并且可以判断引物二聚体和非特异性结合是否会影响实验结果。
当熔解曲线为单峰,认为没有非特异性荧光产物的存在,这样可以得到准确的定量结果。如果体系中存在双峰,并且杂峰的高度大于主峰20%,就认为体系中存在比较严重的非特异性荧光产物和引物二聚体,这样的定量结果就是不准确的。
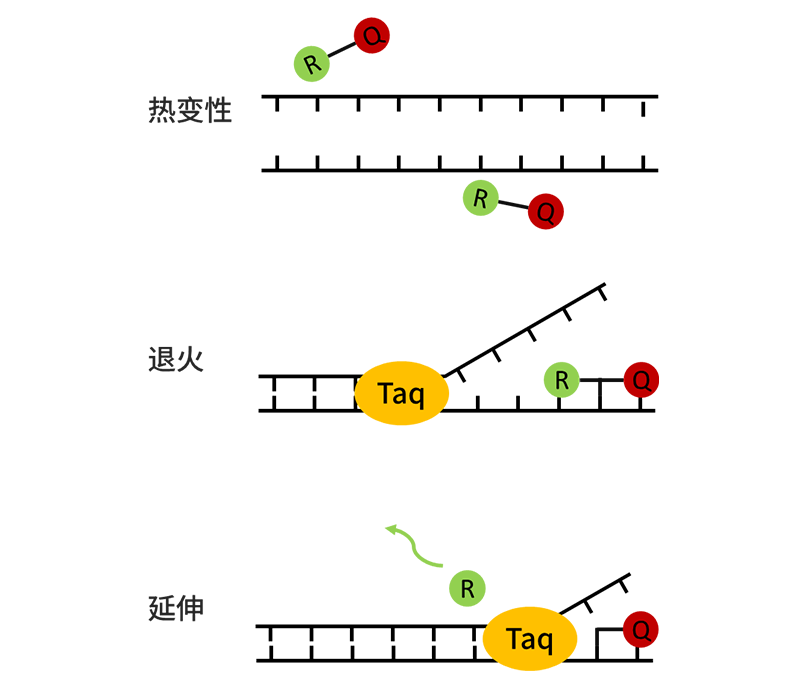
Taqman探针法
介绍这种方法前,首先我们要先了解一下Taqman水解探针的结构,它由5’端荧光报告基团和3’端淬灭基团以及与靶基因特异性结合的序列构成。探针完整时,报告基团发射的荧光信号被淬灭基团吸收,此时仪器不会检测到荧光信号。

当PCR扩增时,由于Taq酶的5’→3’核酸外切酶活性,可以将探针水解,使得荧光报告基团和淬灭基团分离,从而检测到荧光信号。每扩增一条DNA链,就有一个荧光分子形成,荧光信号也就越强。
2、数学原理
荧光信号的强弱代表体系中双链DNA的多少,如果用荧光信号对体系内的PCR过程进行实时监测的话,荧光强度会呈现出一条S型趋势,分为四个时期:基线期、指数扩增期、线性增长期、平台期。
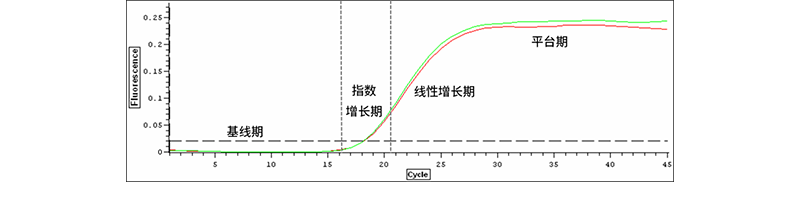
前15个循环,产物比较少,荧光信号弱,大多是背景的荧光信号。当荧光信号超过一定阈值,扩增进入指数期时,这时我们才认为是产物的荧光信号。
什么是荧光阈值:荧光阈值的缺省设置是3-15个循环的荧光信号的标准偏差的10倍。也可手动设置:大于荧光背景值和阴性对照的荧光最高值。一般我们荧光信号达到设定阈值时的扩增循环数,称为Ct值。
Ct值定量的数学原理
理想条件下的PCR反应:×
Xn=Xo× 2n
实际上并不是每次扩增都会翻倍,也就是实际扩增效率不到100%,此时非理想条件下的PCR反应:Xn=Xo(1+Ex)n
n:扩增反应的循环次数;Xn:第n次循环后的产物量;X0:初始模板量;Ex:扩增效率
方程式两边同时取对数得:log Xn=log(Xo(1+Ex)n)
整理方程式得:log Xo=(-log(1+Ex))×n+log Xn
当扩增产物的荧光信号达到荧光阈值时,所经历的循环数为Ct,将其带入方程可得:log Xo=(-log(1+Ex))×Ct+log Xct
可以看出,初始模板量X0的对数与Ct值是呈线性相关的。
初始模板的量越多,达到荧光阈值所需要的循环数就越少,即Ct值越小。所以我们利用几个已知浓度模板和未知浓度的模板一起做相同的反应,利用已知浓度模板的Ct和浓度,拟合Ct与初始模板量的标准曲线,即可读出未知模板Ct值的起始模板量。从线性关系的方程式,可以得到斜率log(1+Ex),进而可以得出反应的扩增效率。最后我们再来看一下,为什么说普通PCR反应无法用于定量呢?
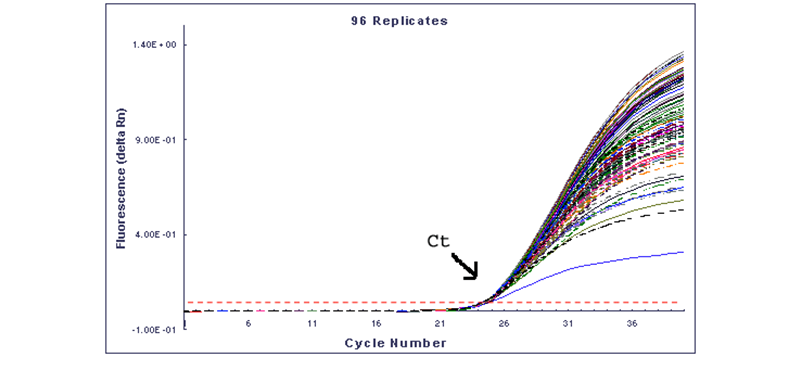
同样的样本在同样的条件下扩增96次得到的曲线图,可以看出96次实验的荧光终点值是不同,但Ct是几乎相同的,所以荧光定量PCR选择用Ct值进行定量。
Azenta安升达可提供荧光定量PCR服务,可以用于绝对定量的测定,比如基因拷贝数的检测等,或者用于相对定量,比如基因表达分析等。
